

“TGF-β/Smad Pathway and how to attenuate tissue fibrosis”
Autor: Dr. León Martin J.
martinjleon87@gmail.com – +54 9 381 5762382
RESUMEN
Las cicatrices aberrantes de la piel, incluidos los queloides y las cicatrices hipertróficas, se caracterizan por la formación y deposición excesivas de colágeno, es el resultado final del proceso patológico de cicatrización de heridas. La vía de señalización TGF-β/Smad en los fibroblastos y miofibroblastos, está involucrada en el proceso de cicatrización de la fibrosis cutánea, la misma es una respuesta de cicatrización de heridas a una lesión celular aguda o crónica que se caracteriza por la acumulación de matriz extracelular (ECM). La evidencia excesiva demostró que la fibrogénesis está asociada con el sistema renina-angiotensina, la inflamación y el estrés oxidativo, la vía de señalización del factor de crecimiento transformador β (TGF-β)/Smad, la vía de señalización Wnt/β-catenina y el metabolismo de los lípidos. Entre ellos, la señalización TGF-β/Smad juega un papel importante en la fibrosis. El TGF-β se encuentra en la matriz en un estado latente y debe activarse antes de que pueda unirse a sus receptores. En los últimos años, se ha prestado más atención a la vía de señalización TGF-β/Smad como objetivo eficaz de la terapia antifibrótica, que, además, es la vía más canónica a través de la cual se regula la formación de colágeno en los fibroblastos y miofibroblastos. Hay dos categorías de estrategias terapéuticas que tienen como objetivo dirigirse a la vía de señalización TGF-β/Smad en fibroblastos y miofibroblastos para interferir con sus funciones celulares y reducir la proliferación celular. La primera estrategia terapéutica incluye medicamentos, y la segunda estrategia se compone de terapia genética y celular. Por lo tanto, el objetivo de esta revisión es evaluar críticamente estas dos estrategias terapéuticas principales que se dirigen a la vía TGF-β/Smad para atenuar la formación anormal de cicatrices en la piel.
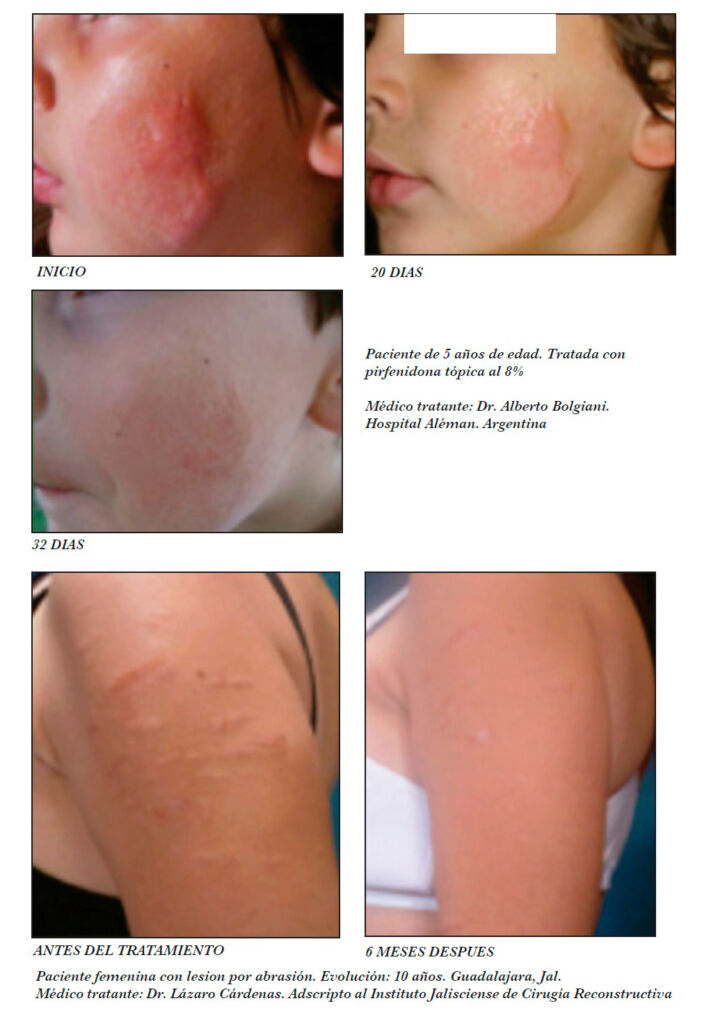
El uso de gel tópico de Pirfenidona a una concentración del 8% se prescribe comúnmente para el tratamiento de heridas como las cirugías, quemaduras, dehiscencias y úlceras. Se aplica de manera tópica sobre las zonas a tratar, siendo menores los efectos adversos informados que con la administración sistémica. La frecuencia de aplicación es dos veces por día en forma de capa delgada y la duración del tratamiento puede variar según la gravedad de las cicatrices y la respuesta individual al medicamento.
Palabras Clave: Cicatriz hipertrófica, queloide, fibroblastos, TGF-β/Smad, estrategias terapéuticas, pirfenidona.
ABSTRACT
Aberrant skin scars, including keloids and hypertrophic scars, are characterized by excessive collagen formation and deposition, is the end result of the pathological process of wound healing. The TGF-β/Smad signaling pathway in fibroblasts and myofibroblasts is involved in the healing process of skin fibrosis, which is a wound-healing response to acute or chronic cell injury characterized by matrix accumulation extracellular (ECM). Excessive evidence demonstrated that fibrogenesis is associated with the renin-angiotensin system, inflammation and oxidative stress, the transforming growth factor-β (TGF-β)/Smad signaling pathway, the Wnt/β-catenin signaling pathway and lipid metabolism. Among them, TGF-β/Smad signaling plays an important role in fibrosis. TGF-β is in the matrix in a latent state and must be activated before it can bind to its receptors. In recent years, more attention has been paid to the TGF-β/Smad signaling pathway as an effective target of antifibrotic therapy, which is also the most canonical pathway through which collagen formation in fibroblasts is regulated. and myofibroblasts. There are two categories of therapeutic strategies that aim to target the TGF-β/Smad signaling pathway in fibroblasts and myofibroblasts to interfere with their cellular functions and reduce cell proliferation. The first therapeutic strategy includes drugs, and the second strategy consists of gene and cell therapy. Therefore, the aim of this review is to critically evaluate these two main therapeutic strategies that target the TGF-β/Smad pathway to attenuate abnormal skin scar formation.
The use of topical Pirfenidone gel at a concentration of 8% is commonly prescribed for the treatment of wounds such as surgeries, burns, dehiscence and ulcers. It is applied topically to the areas to be treated, with fewer reported adverse effects than with systemic administration. The frequency of application is 2 times a day in the form of a thin layer and the duration of treatment may vary depending on the severity of the scars and the individual response to the medication.
Keywords: Hypertrophic Scar; Keloid; Fibroblast; TGF-β/Smad; Therapeutic strategies, pirfenidone.
MATERIALES Y MÉTODOS
Se realizó una búsqueda bibliográfica en Pubmed utilizando palabras clave Hypertrophic Scar; Keloid; Fibroblast; TGF-β/Smad; Therapeutic strategies, pirfenidone, aplicando un filtro de 5 años, donde se encontraron un total de 25 artículos, de los cuales 10 de ellos tienen relación con fibrosis tisular, vías de señalización TGF-β/Smad y el uso de la pirfenidona como antifibrótico, el resto se excluyó por estar relacionado solamente con fibrosis hepática, renal, carcinogénesis y temas no afines a la fibrosis tisular. Además, se realizo una evaluación retro prospectiva de pacientes del CEPAQ (Hospital Aleman) que se incluyen en protocolo de investigación de México, a cargo del Dr. Bolgiani, que prueba el uso de la Pirfenidona tópica como agente antifibrótico en heridas agudas y crónicas.
OBJETIVOS
El objetivo de esta revisión es evaluar críticamente estas dos estrategias terapéuticas principales que se dirigen a la vía TGF-β/Smad para atenuar la formación anormal de cicatrices en la piel, y el uso tópico de la Pirfenidona al 8% como modulador del TGF-β/.
INTRODUCCIÓN
La piel es un órgano del sistema tegumentario que consta de múltiples capas de tejido ectodérmico que actúa como una barrera externa suave para cubrir los músculos, huesos, nervios y órganos internos subyacentes y es la primera línea de defensa contra factores externos. Hay muchos estímulos externos destructivos que pueden provocar lesiones cutáneas en la piel, como traumatismos, quemaduras y cirugía. La formación de cicatrices ocurre como resultado de una lesión en la piel y puede conducir a un deterioro estético o una función reducida de la piel y trastornos psicológicos, incluida la autodegradación y la ansiedad (1,2). Por lo tanto, el estudio de los mecanismos moleculares de la formación de cicatrices es importante para desarrollar una mejor comprensión del proceso con el fin de reducir la formación de cicatrices, prevenir la función cutánea deteriorada y mejorar la apariencia de la piel. La formación de cicatrices es un componente del proceso de cicatrización de heridas, que es un proceso fisiológico normal, complejo y estrictamente regulado que involucra la regulación de la migración celular, la inflamación, la inervación y la angiogénesis. Sin embargo, un proceso patológico de cicatrización de heridas puede conducir a la formación de cicatrices desorganizadas, que se caracterizan por una fibrosis excesiva.
Las cicatrices hipertróficas son el resultado del daño tisular y de una respuesta inflamatoria prolongada y fibrótica excesiva, que produce la deposición exorbitante de colágeno durante el proceso de reparación. Son elevadas, rojas, se asocian con dolor intermitente, picazón persistente y sensación de contracción. Estas cicatrices patológicas pueden provocar un deterioro funcional grave y morbilidad psicológica. Las principales células efectoras son los fibroblastos, que se asocian con una inflamación excesiva en la dermis reticular y el depósito de proteínas de la matriz extracelular junto con niveles elevados de factor de necrosis tumoral α (TNF-α), interleucina-6 (IL-6) y factor de crecimiento transformante b1 (TGF-b1). La pirfenidona (Pf) es un fármaco de molécula pequeña aprobado por la FDA indicado para la fibrosis pulmonar idiopática y se están realizando estudios para reutilizar el fármaco para otras enfermedades fibróticas (3). La Pf se aplica tópicamente para prevenir y mitigar las cicatrices hipertróficas asociadas a quemaduras. La Pf tópica existe comercialmente en México en formato de gel (4).
Las cicatrices hipertróficas pueden ocurrir hasta en el 91% de todas las lesiones por quemaduras y entre el 40-70% de las cirugías (5). Hay estudios que demuestran la eficacia de la pirfenidona en tratamientos de forma tópica, como el realizado en ratones con quemaduras profundas de espesor parcial (6). Y en pacientes humanos para mejorar la epitelización de sitios donantes de injertos de piel de espesor parcial (STSG) (7, 8). La administración tópica de Pf puede considerarse como un uso terapéutico cuando se requieren tiempos de curación más rápidos, como en las zonas de donantes en pacientes quemados.
La cicatrización anormal en la piel incluye la formación de dos tipos diferentes de cicatrices, queloides e hipertróficas, que tienen diferentes características clínicas. Una cicatriz hipertrófica es una cicatriz ancha, engrosada y, a menudo, elevada que se desarrolla en el sitio de la lesión cutánea que puede causar picazón y, por lo general, no se extiende más allá del límite de la herida original. Una de las principales causas de formación de cicatrices hipertróficas es la tensión mecánica en la herida (9), pero las cicatrices hipertróficas suelen sufrir una regresión espontánea en uno o dos años. Las cicatrices queloides son lesiones gomosas duras o nódulos fibrosos brillantes asociados con prurito y dolor. Los queloides se consideran tumores benignos, pero a menudo se identifican erróneamente como tumores malignos debido a su apariencia clínica. Son crecimientos amorfos elevados que se extienden más allá del límite del sitio original de la lesión cutánea e invaden los tejidos adyacentes. Las personas con cualquier nivel de traumatismo cutáneo, incluyendo cirugía, piercings, acné, tatuajes, picaduras de insectos, quemaduras, laceraciones, abrasiones, vacunas y cualquier otro proceso que resulte en inflamación cutánea, están predispuestas al desarrollo de cicatrices hipertróficas y queloides (10,11). Ambos tipos de cicatrices se asocian con una fibrosis excesiva causada por la sobreproducción y el depósito de un exceso de matriz extracelular (MEC), compuesta principalmente de colágeno, con cicatrices hipertróficas que tienen niveles elevados de colágeno tipo III paralelo y cicatrices queloides que tienen un exceso de colágeno tipo I y III desorganizado (11). La fibrosis patológica puede ocurrir sin la presencia de una herida real siempre que haya una respuesta inflamatoria en el tejido. Esta respuesta inflamatoria puede promover un estado proliferativo en los fibroblastos, que puede iniciarse y mantenerse el tiempo suficiente para dar lugar a un posterior depósito continuo de MEC. Por ejemplo, la cirrosis hepática puede ser el resultado de estímulos inflamatorios crónicos, como los virus (12). Por lo tanto, podemos definir la cicatrización o fibrosis como una característica patológica de la mayoría de las enfermedades inflamatorias crónicas con acumulación de componentes de la matriz extracelular en exceso, que son producidos por fibroblastos y miofibroblastos (13).
La desregulación de la señalización de TGF-β / Smad es un factor importante en el marco de la cicatrización y la fibrosis, lo que conduce a una síntesis y depósito de colágeno aberrante, una mayor proporción de colágeno I / III y la formación de haces de fibras de colágeno anormalmente reticulados (14,15,16). El TGF-β juega un papel fundamental en la producción del fenotipo de miofibroblastos que es responsable de la deposición y contracción masiva de colágeno de las heridas (17). El factor de crecimiento transformante (TGF) fue identificado por primera vez por De Larco y Todaro en 1978, llamado así porque posee la capacidad de inducir un fenotipo transformado en células no transformadas. No fue hasta 1986 que Roberts et al. identificaron que el TGF puede estimular la producción de ECM en los fibroblastos. Border et al. comenzaron el estudio de la relación entre TGF y glomerulonefritis en 1990 a través de su investigación de TGF y fibrosis. El estudio de los Smads como efectores del TGF-β y los aspectos clave de los mecanismos de señalización generales se estudiaron ampliamente entre 1995 y 1996 (18).
En general, la formación de cicatrices y la fibrosis, como la cirrosis después de una infección viral y las placas fibrosas formadas después de un infarto de miocardio, siguen un proceso predecible. Sin embargo, ciertas enfermedades fibróticas suelen tener un proceso prolongado que puede tardar décadas en desarrollarse. A menudo, este proceso no se identifica hasta que el paciente tiene síntomas clínicos y busca atención médica. Se necesitan tratamientos para la fibrosis y profilaxis de la fibrosis no formada. Para las personas que tienen formación anormal de cicatrices, como cicatrices queloides, se ha sugerido una variedad de terapias, que incluyen tratamientos quirúrgicos, no quirúrgicos y de modalidad combinada, que involucran esteroides intralesionales o tópicos, crioterapia, escisión quirúrgica, radioterapia y terapia con láser (10). Estas terapias tienen una alta tasa de éxito y una baja tasa de recurrencia. Sin embargo, aún falta un consenso con respecto al gold estandar para mejorar de forma permanente la cicatrización patológica, debido a los criterios de valoración mal definidos en el tratamiento, el seguimiento inadecuado y la falta de estudios prospectivos. Además, algunos tratamientos se acompañan de efectos secundarios inevitables, como el uso de inyección intralesional de corticosteroides, que pueden provocar dolor y atrofia de los tejidos normales circundantes, especialmente osteoporosis (19). El desarrollo de terapias para la reparación de tejidos normales y patológicos ha sido un desafío debido al extenso desequilibrio e interconexión entre las vías de señalización que subyacen a estas patologías, así como a la dificultad inherente para determinar con precisión las vías que se ven afectadas en cada caso. Como se conoce bien la vía de señalización canónica TGF-β / Smad involucrada en el proceso de cicatrización de la fibrosis cutánea, muchos esfuerzos de investigación se han centrado en apuntar a la señalización TGF-β / Smad en fibroblastos y miofibroblastos. Un enfoque importante de la presente revisión es evaluar críticamente los roles de las dos categorías principales de estrategias terapéuticas que se han desarrollado en los últimos cinco años que se dirigen a la vía de señalización TGF-β / Smad para atenuar la formación anormal de cicatrices en la piel. Las dos categorías de estrategias terapéuticas incluyen estrategias terapéuticas medicamentosas (Tabla 2) y estrategias terapéuticas genéticas y celulares (Tabla 3). Estas estrategias terapéuticas se dirigen principalmente a la vía de señalización TGF-β/Smad en fibroblastos y miofibroblastos para interferir con sus funciones y reducir la proliferación celular.
El TGF-β1 se considera un mediador crucial en la fibrosis tisular y causa cicatrices tisulares en gran medida al activar a su señalización decapentapléjica (Smad). TGF-β1 activa directamente la señalización de Smad, lo que desencadena la sobreexpresión del gen profibrótico. Estudios excesivos han demostrado que la desregulación de la vía TGF-β1/Smad era un mecanismo patógeno importante en la fibrosis tisular. Smad2 y Smad3 son los dos principales reguladores que promueven la fibrosis tisular mediada por TGF-β1, mientras que Smad7 sirve como un regulador de retroalimentación negativa de la vía TGF-β1/Smad, lo que protege contra la fibrosis.
TGF-β es un mediador multifuncional que regula la proliferación, la diferenciación, la apoptosis, la adhesión y la migración en varias células, como los macrófagos, las células T y B activadas, las células hematopoyéticas inmaduras, los neutrófilos y las células dendríticas. Hay tres isoformas de TGF-β, incluyendo el factor de crecimiento transformador β1 (TGF-β1), el factor de crecimiento transformador β2 (TGF-β2) y el factor de crecimiento transformador β3 (TGF-β3). TGF-β1 se expresa en células de tejido endotelial, hematopoyético y conectivo, y TGF-β2 se expresa en células epiteliales y neuronales, mientras que TGF-β3 se expresa principalmente en células mesenquimales.
El bloqueo de TGF-β neutralizando los anticuerpos TGF-β, la decorina y los oligonucleótidos previene o mejora la fibrosis. La decorina es un proteoglicano pequeño, puede ser celular o de ECM se caracteriza por poseer una región rica en leucinas con una cadena de glicosaminoglicanos que bien puede ser del tipo condroitín sulfato o bien del tipo dermatán sulfato.
La expresión génica de TGF-β1, smad2 y smad3 podría detectarse en la piel fetal y después del nacimiento. En la piel del feto gestacional temprano, las expresiones genéticas de TGF-β1y smad2 son débiles. Junto con el avance de la edad gestacional, la expresión génica en la piel se hace cada vez más fuerte. En la del feto gestacional tardío y en la piel después del nacimiento, el contenido de transcripción de estos dos genes aumenta significativamente en comparación con el feto gestacional temprano. Por el contrario, la expresión génica de smad3 fue aparentemente más alta en la piel fetal más joven en comparación con la de los ancianos y en comparación con la piel fetal tardía.
La vía de señalización mediada por TGF-β1 podría estar involucrada en la regulación del desarrollo de la piel en la etapa embrionaria y también en la cicatrización de heridas después del nacimiento. La relativa falta de expresión de los genes TGF-β1y smad2 en pieles de fetos más jóvenes podría contribuir a la curación sin cicatrices.
El proceso normal de cicatrización de heridas
La formación anormal de cicatrices se puede prevenir mediante un proceso eficaz de cicatrización de heridas, pero la inflamación prolongada o persistente en este proceso puede exacerbar la fibrosis de la piel. Por lo tanto, comprender el proceso normal de cicatrización de heridas es crucial para la identificación de la cicatrización patológica de heridas y la identificación de posibles terapias. Describimos las etapas del proceso normal de cicatrización de heridas en la Tabla 1. La Fig. 1 ilustra el proceso normal de cicatrización de heridas que conduce a la formación de cicatrices. El proceso de cicatrización de heridas se divide en fases de hemostasia, inflamación, proliferación y remodelación, y para una cicatrización exitosa de heridas, todos los procesos deben ocurrir en la secuencia y el marco de tiempo adecuados. El sangrado debido a un traumatismo conduce a la vasoconstricción local y la agregación de plaquetas para formar un tapón para llegar a la hemostasia primaria, lo que en consecuencia activa la cascada de coagulación y da como resultado la formación de coágulos de fibrina para formar la hemostasia secundaria (20).
Durante el proceso de hemostasia, las citocinas proinflamatorias y los factores de crecimiento, como el factor de crecimiento derivado de plaquetas (PDGF), el factor de crecimiento epidérmico (EGF), el factor de crecimiento de fibroblastos 2 (FGF 2; también conocido como bFGF), la interleucina 8 (IL-8) y el TGF-β, se liberan del coágulo recién formado y los tejidos dañados circundantes (21).
La siguiente fase es la fase inflamatoria, en la que las células inflamatorias ingresan al área de la herida desde el sistema inmunológico debido a la señalización de quimiocinas, que se caracteriza por la infiltración secuencial de neutrófilos, macrófagos y linfocitos. La abundancia de neutrófilos no solo proporciona una defensa crucial contra los microbios invasores, sino que también elimina tanto el patógeno como los restos de tejido en el área de la herida. Esto ocurre a través de la fagocitosis por la liberación de especies de nitrógeno y especies reactivas de oxígeno (ROS) (22). Los neutrófilos también son esenciales para el reclutamiento de otras células inflamatorias, por ejemplo, macrófagos derivados de monocitos. Los macrófagos derivados de monocitos se diferencian en macrófagos maduros de heridas debido a la influencia de citocinas locales, como el interferón-γ (IFN-γ) y productos bacterianos, como el lipopolisacárido. Los macrófagos maduros continúan el proceso de eliminación fagocitando restos de tejidos, organismos microbianos y células apoptóticas, y estos macrófagos con propiedades antimicrobianas se consideran el tipo de macrófago M1 (23). Después de eliminar las células senescentes y defenderse de posibles patógenos, varios macrófagos M1 sufren apoptosis. Los pocos macrófagos restantes se convierten en el tipo de macrófago M2 como consecuencia de los cambios en la expresión de citoquinas, por ejemplo, IL-13 e IL-4 (24).
Los macrófagos M2 realizan diferentes funciones en el proceso de cicatrización de heridas, ya que son responsables de la transición a la fase proliferativa del proceso de cicatrización de heridas. Los macrófagos M2 liberan factores de crecimiento, como el factor de crecimiento endotelial vascular (VEGF), PDGF y TGF-β, que contribuyen al cambio a la fase proliferativa (20). Estos factores promueven la angiogénesis, la reepitelización y la producción de colágeno (25). Durante la fase proliferativa, los fibroblastos son reclutados y comienzan a producir colágeno, glicosaminoglicanos, proteoglicanos, fibronectina y elastina, que son los principales componentes de la ECM (21,26). Los fibroblastos también experimentan proliferación, migración y diferenciación debido a la presencia de factores de crecimiento, como FGF, PDGF y TGF-β. Los fibroblastos y las células endoteliales son esenciales para el crecimiento capilar y la formación de tejido de granulación en el sitio de la herida en la dermis reparadora (21). La contracción de la herida es un proceso crucial en la cicatrización de heridas que ocurre a través de la acción de los miofibroblastos, que son células contráctiles especializadas caracterizadas por la expresión de actina del músculo liso α (α-SMA) (27,28). Estas células desaparecen gradualmente del tejido de granulación a medida que la herida madura. En comparación con los fibroblastos dérmicos normales, los miofibroblastos producen mayores cantidades de componentes de ECM (28). Curiosamente, las fuentes de miofibroblastos se remontan a diferentes células, incluidas las células epiteliales de la piel, los pericitos y las células del músculo liso vascular pulmonar. Sin embargo, la principal fuente de miofibroblastos es la transición o activación de fibroblastos residentes en respuesta a varios desencadenantes, incluido el TGF-β1 (29).
Después de la proliferación y la síntesis de ECM, la cicatrización de heridas pasa a la fase final de remodelación más larga, que es un equilibrio entre la producción, descomposición y remodelación de ECM, que continúa durante semanas o meses e incluso, en algunos casos, durante años (30). Esta fase se caracteriza por una proliferación celular reducida, apoptosis de la mayoría de las células endoteliales, macrófagos y miofibroblastos, reemplazo de glicosaminoglicanos por proteoglicanos y sustitución de colágeno III por colágeno I a través de la acción de colagenasas y metaloproteasas de matriz (MMP) (30,31). El resultado crítico de la reorganización de la ECM es remodelar para formar una arquitectura que se asemeje más a los tejidos normales. La manifestación clínica de esta fase es una cicatriz blanda palpable, que varía de color rosado a pálido debido a la microvasculatura de la cicatriz (32). A medida que la vascularización retrocede, el tono de la cicatriz cambia en consecuencia.
Función de la vía dependiente de Smad inducida por TGF-β
La formación de cicatrices es un sistema de reparación eficiente que es importante para la evolución genética de los seres humanos. Sin embargo, cuando una cicatriz recién formada ocupa una gran parte del tejido normal, puede provocar diversos grados de desfiguración y deterioro funcional. Por ejemplo, después de un infarto de miocardio, la necrosis tisular aguda y la lesión por reperfusión conducen a la cicatrización del corazón, lo que resulta en la alteración del tejido normal del corazón y ritmos cardíacos anormales (arritmias) debido a la alteración grave de la conductancia de la señal eléctrica miocárdica (33). Independientemente del origen de las cicatrices fibrosas, ya sea la formación de cicatrices en la superficie de la piel o la fibrosis de los órganos internos, todas las enfermedades fibróticas están asociadas con la activación de los miofibroblastos productores de MEC, que son los mediadores clave de la remodelación del tejido fibrótico (34, 35). La señalización aberrante de TGF-β/Smad en miofibroblastos se asocia con la formación de cicatrices patológicas, como la formación de cicatrices hipertróficas.
La síntesis de colágeno y α-SMA inducida por TGF-β juega un papel crucial en la reparación de tejidos y el desarrollo de fibrosis en fibroblastos y miofibroblastos (17, 36). Utilizamos α-SMA y colágeno como ejemplos para arrojar luz sobre los mecanismos de transcripción inducidos por la vía de señalización TGF-β/Smad. TGF-β es una citocina que se expresa de forma ubicua durante el proceso de cicatrización de heridas y puede ser sintetizada por tejidos dañados, fibroblastos y macrófagos M2 (20, 21). El TGF-β pertenece a una gran superfamilia que contiene factores de crecimiento polipeptídicos estructuralmente relacionados, que incluyen ganglionares, activinas, proteínas morfogenéticas óseas (BMP), inhibinas y factores de crecimiento y diferenciación (GDF) (37, 38). TGF-β normalmente funciona para modular numerosos aspectos fundamentales de la homeostasis celular, incluido el crecimiento celular, la migración, la apoptosis, la diferenciación y la producción de colágeno (39, 40). El ligando TGF-β inicia señales a través del ensamblaje de un complejo serina/treonina quinasa que está compuesto por un receptor de tipo I (activina similar a la quinasa (ALK) 1-7) y un receptor de tipo II en la superficie celular. La unión del ligando y el receptor da como resultado la activación del receptor tipo II, que luego fosforila el receptor tipo I. Además, se ha identificado un número creciente de receptores tipo III, que actúan como correceptores que se unen al TGF y regulan la señalización del TGF-β (41). Por ejemplo, los receptores de tipo III pueden unirse a ligandos como los ligandos TGF-β (42), BMP (43) e inhibina (44, 45) para modular la función, mediar la interacción de estos ligandos con otros receptores de la superfamilia TGF-β y regular la localización de estos receptores (41, 42).
Hay ocho madres pequeñas diferentes contra las proteínas decapentapléjicas (Smad) que conforman tres categorías funcionales: el Co-mediador Smad (Co-Smad), el Smad regulado por el receptor (R-Smad) y el inhibidor Smad (I-Smad) (46). Después de la fosforilación, el receptor tipo I reconoce y fosforila específicamente los R-Smads, incluidos Smad2 y Smad3. La fosforilación desestabiliza la interacción con el anclaje Smad para la activación del receptor (SARA) y aumenta la afinidad por Smad4 (el llamado Co-Smad) (46). Cuando Smad2 y Smad3 se liberan de SARA, pueden interactuar con Smad4 para formar un complejo transcripcional, que es libre de translocarse al núcleo, unirse a coactivadores o correpresores transcripcionales y regular la actividad transcripcional de varios genes (46). Por ejemplo, el dominio de homología Mad 1 de Smad3 y Smad4 media la unión directa a los elementos de unión a Smad3 en la parte superior del promotor α-SMA del núcleo de 125 pb, regulando así la activación transcripcional del gen α-SMA (47). La secuencia CAGACA está presente en la región de respuesta TGF-β1 del promotor del gen del colágeno tipo I, actuando como un elemento funcional de unión a Smad en el promotor del gen inhibidor del activador del plasminógeno-1 (48, 49). Smad3 interactúa con los coactivadores transcripcionales ubicuos p300 y la proteína de unión a CREB y se une a la secuencia CAGACA, estimulando la transcripción del gen del colágeno tipo I (50).
Además, Smad2 y Smad3 son reconocidos por los receptores de activina y ganglionar estrechamente relacionados. Otros R-Smads como Smad1, Smad5 y Smad8 son reconocidos principalmente por BMP (46). Una vez que se activa la vía Smad inducida por TGF-β, se activan numerosos mecanismos de retroalimentación para modular la duración de la señalización (51). Los I-Smads (Smad6 y Smad7), que están regulados por la señalización de TGF-β y BMP, inhiben la señalización de la superfamilia TGF-β al interferir directamente con la asociación entre R-Smads y sus respectivos receptores tipo I, lo que lleva a una fosforilación reducida de R-Smads (52,53). El mecanismo para desactivar la señalización de TGF-β implica la ubiquitinación de Smads en el núcleo y el citoplasma, seguida de la degradación mediada por proteasoma de las proteínas Smad (54). Hay tres tipos principales de ubiquitinaligasas E3 que median la ubiquitinación de Smads. El factor 1 relacionado con la ubiquitina Smad (Smurf1) interactúa selectivamente con los Smads 1, 5 y 8 en el citoplasma para desencadenar su ubiquitinación (55), y Smurf2 se dirige selectivamente a Smad2 activado en el núcleo para su degradación (56). La ubiquitinación de Smad3 en el núcleo está mediada por un complejo diferente de ubiquitinaligasa E3, ROC1-SCFFbw1a, que consta de ROC1, Skp1, Cullin1 y Fbw1a (57). Cabe destacar que tanto Smurf1 como Smurf2 pueden ser reclutados por Smad7 y formar un complejo estable con Smad7 para mediar la ubiquitinación del receptor TGF-β tipo I, con Smad7 experimentando ubiquitinación y degradación durante este proceso (58,59,60,61). En la Fig. 2 se muestra el mapa de la vía dependiente de Smad inducida por TGF-β.
Modulación de la vía dependiente de smad inducida por TGF-β
Dado que la vía dependiente de Smad inducida por TGF-β es fundamental para la pathogenesis de todas las enfermedades fibróticas, se han investigado varias estrategias terapéuticas para apuntar a la vía de señalización para atenuar la formación aberrante de cicatrices cutáneas. Estas estrategias se pueden subdividir en estrategias terapéuticas de medicamentos, posibles estrategias terapéuticas genéticas y celulares. Los efectos de ambas estrategias terapéuticas se muestran en la Fig.1, Fig.3.
Estrategias terapéuticas de medicamentos
Angiogenina es una molécula potente que induce el crecimiento tumoral al promover la neovascularización, la sobreexpresión de angiogenina se detecta en heridas por quemaduras profundas de espesor parcial (62, 63) y también se ha demostrado que actúa como un agente neuroprotector en las neuronas motoras en la esclerosis lateral amiotrófica (ELA) (64). Es importante destacar que la angiogenina está involucrada en las respuestas inflamatorias y proliferativas durante el proceso de cicatrización de heridas, ya que los fibroblastos cicatriciales producen angiogenina de manera dependiente del tiempo (65). Estos hallazgos sugieren que la angiogenina juega un papel en la promoción de fibrosis en la piel en condiciones patológicas y que los altos niveles de proteína angiogenina exógena pueden influir significativamente en la función biológica y la proliferación de fibroblastos cicatriciales en pacientes quemados in vitro a través de la inhibición de la señalización TGF-β1/Smad2, disminuyendo la proliferación de fibroblastos cicatriciales y la secreción de TGF-β1 (61). A pesar de los efectos positivos del uso de la angiogenina para los fibroblastos en la cicatriz hipertrófica de pacientes quemados y su papel protector en las neuronas, no está claro si esta molécula seria lo suficientemente segura para su uso en la clínica, ya que puede provocar un crecimiento tumoral subyacente.
Baicaleína o 5,6,7-trihidroxiflavona es un tipo de flavonoide extraído originalmente de las raíces de la hierva Scutellaria Baicalensis y Scutellaria Lateriflora. Se ha demostrado que el extracto inhibe la proliferación y metástasis de las células del carcinoma hepatocelular (66) y actúa como un potente agente neuroprotector en las enfermedades de Alzheimer y Parkinson (67,68). La baicaleína también puede suprimir la progresión tumoral al inhibir el crecimiento de células tumorales y la angiogénesis tumoral (69) atenuar las actividades fibróticas en los fibroblastos cardiacos e inhibir la migración de células estrelladas hepáticas en la fibrosis hepática (70,71). Además, posee efectos antiinflamatorios a través de la inhibición de la transactivación de NF-kB (72). Curiosamente, también es una terapia prometedora para la antifibrogénesis en la piel ya que ejerce un efecto inhibitorio sobre la vía de señalización TGF-β/Smad2/3 en fibroblastos tanto in vivo como in vitro. Suprime significativamente la proliferación y activación de fibroblastos derivados de la cicatriz hipertrófica e inhibe la expresión de α-SMA. Esto conduce a una alteración de las capacidades contráctiles y de migración de los fibroblastos derivados de cicatrices hipertróficas, lo que disminuye la deposición de colágeno y atenúa la formación de cicatrices hipertróficas (73,74). Los mecanismos subyacentes de la baicaleína pueden derivarse de su unión selectiva con la ATP quinasa similar al receptor de activina5 (ALKS), un receptor de TGF-β I, sin ejercer ningún cambio en la expresión de los receptores I/II de Smad6 y Smad7. La formación de cicatrices puede mejorarse con la aplicación de baicaleína. Sin embargo, se necesita más investigación para comprender mejor los mecanismos moleculares y los posibles efectos secundarios.
Ginsenósidos Rg3 (Rg3). Es una sustancia extraída de una hierba tradicional china, el Panax Ginseng. Se ha informado que Rg3 exhibe numerosos efectos antitumorales, incluida la inhibición de la proteína del cáncer colorrectal a través de la regulación de las vías de señalización Wnt/βcatenina y C/EBPβ/NF-kB (75, 76). Además también se ha demostrado que Rg3 promueve la apoptosis en el cáncer de ovario humano a través de la regulación de las proteínas de la familia fosfatidilinositol 3-quinasa (PI3K)/Akt (77). Rg3 parece ser un agente terapéutico potencial que se dirige a los queloides similares a tumores. Se ha demostrado que Rg3 suprime la proliferación de fibroblastos queloides al disminuir notablemente la expresión del marcador de proliferación Ki-67 de una manera dosis dependiente (78). Rg3 bloquea la invasión de fibroblastos queloides a través de la regulación de los niveles de expresión de ARNm de MMP-1 y MMP-3, que se expresan en gran medida en los queloides, lo que lleva a la degradación de la MEC (78, 79, 80). Además, se ha demostrado que inhibe la angiogénesis en cultivos de explantes queloides al reducir notablemente los niveles de expresión de ARNm de VEGF (78). Rg3 también reprime la migración de fibroblastos queloides a través de la regulación positiva de la expresión de genes antifibróticos, incluidos INF-g y TGF-β3, y la regulación negativa de la expresión de genes profibróticos, incluidos el a-SMA y el factor de crecimiento de tejido conectivo (CTGF). En resumen, Rg3 tiene un efecto inhibitorio sobre las vías de señalización TGF-β/Smad y aumenta los niveles de expresión de ARNm de Smad7 (78). Estos hallazgos sugieren que Rg3 puede ser una terapia potencial para la inhibición de la formación de queloides. Sin embargo, se ha demostrado que induce la apoptosis de los fibroblastos queloides de una manera dosis dependiente. Se necesitan estudios futuros para determinar si Rg3 causa daño en las células normales, ya que esto puede conducir a problemas de seguridad para el uso clínico.
Kaempferol es una molécula flavonoide, natural que se aísla originalmente del té, el brócoli y otras fuentes vegetales y se ha demostrado que posee una variedad de actividades, incluida la antiinflamación (81) y la inhibición del crecimiento tumoral (82). Puede proporcionar protección contra el daño cutáneo inducido por los rayos ultravioleta a través de la actividad antioxidante y la absorción directa de la luz (83). Se ha demostrado que el kaempferol inhibe significativamente la formación de cicatrices hipertróficas en un modelo de ratón inducido a través de la atenuación de la síntesis de colágeno y la supresión de la proliferación y activación de fibroblastos cicatriciales hipertróficos humanos in vitro. Esto se debe a la unión selectiva del kaempferol al receptor tipo I del TGF-β, lo que lleva a la inactivación del receptor tipoI. Esto da como resultado la fosforilación reducida de Smad2/3 y la inhibición de la vía de señalización TGF-β1/Smad (84).
Loureirin B es un ingrediente del extracto etanólico de Resina Draconis (RDEE), un tipo de resina roja aislada del tallo del árbol de Dracaenaco chinchinensis, utilizada como medicina tradicional china (85) Se ha demostrado que la loureirina B es eficaz en la profilaxis y el tratamiento de cicatrices cutáneas patologicas. La loureirina B tiene un efecto supresor sobre la expresión de colágeno I, colágeno III y α-SMA en fibroblastos cicatriciales hipertróficos de manera dependiente de la dosis en un modelo de cicatriz de oreja de conejo, lo que conduce a una mejor disposición de las fibras de colágeno y a la inhibición de la diferenciación de miofibroblastos (86). En los fibroblastos normales de piel humana estimulados por TGF-β1, la aplicación de loureirina B regula notablemente la expresión de MMP-1, MMP-3, MMP-9 y MMP-13, que provocan la degradación de la MEC, y suprime eficazmente la vía TGF-β/Smad a través de la interrupción de la fosforilación de Smad2 y Smad3 (86). Tanto los estudios in vivo como in vitro han demostrado que la loureirina B puede ser beneficiosa en la prevención y el tratamiento de cicatrices cutáneas anormales. También se ha demostrado que la loureirina B altera el ciclo celular, lo que lleva a una proliferación bloqueada sin ningún efecto sobre la apoptosis (86). Sin embargo, se necesitan estudios futuros para determinar si la loureirina B tendrá efectos sobre el ciclo celular de otras células, por ejemplo, las células de carcinoma.
Proteína morfogenética ósea-7 (BMP-7) es un miembro de la super familia TGF-β. Sin embargo, las funciones de BMP-7 y TGF-β son muy diferentes. Se ha demostrado que una alta proporción de BMP-7/TGF-β atenúa la fibrosis hepática de rata, lo que sugiere que BMP-7 puede contrarrestar los efectos fibróticos inducidos por TGF-β1 (87). Varios estudios han sugerido que el tratamiento con BMP reduce la fibrosis de los órganos viscerales, incluida la atenuación de la remodelación ventricular adversa (88, 89), la prevención de la fibrosis renal (90) y la atenuación de la fibrosis hepática (91) y la fibrosis pulmonar inducida por sílice (92). También se ha demostrado que BMP-7 afecta la fibrosis de la piel, y a que la inyección de BMP-7 exógeno en el sitio de la lesión térmica en ratones promueve la cicatrización de heridas y la inhibición de la formación de cicatrices. Esto ocurre debido a la activación de la vía de señalización BMP-7/Smads1,5 y 8 a través de aumentos significativos en la fosforilación de Smads1,5 y 8 (93). El tratamiento con BMP-7 también disminuye notablemente la expresión de α-SMA, TGF-β1 y CTGF y reduce los niveles de proteínas de colágeno I y III (93). Además, BMP-7 induce la apoptosis de los fibroblastos cicatriciales, lo que se puede demostrar por la disminución del gen BCL2 antiapoptótico, el aumento del gen Bax proapoptótico y la caspasa-3 (93). En resumen, la aplicación de BMP-7 puede ser relativamente segura. Sin embargo, se necesita más trabajo para determinar si BMP-7 puede inducir la apoptosis de los fibroblastos normales de la piel.
Galangin (3,5,7-trihidroxiflavona) es otro flavonoide activo natural que se extrae originalmente de las raíces de Alpinia officinarum. La galangina tiene múltiples funciones biológicas, incluida la actividad anticancerígena (carcinoma hepato celular (95), retinoblastoma (95), carcinoma laríngeo (96) y osteosarcoma (97)) y la atenuación del estrés oxidativo, como mejorar la nefrotoxicidad (98) inducida por cisplatino y suprimir el envejecimiento inducido por peróxido de hidrógeno (H2O2) en fibroblastos dérmicos humanos (99). También se ha demostrado que la galangina tiene una actividad antiinflamatoria (100). La galangina también puede inhibir significativamente la producción de colágeno e inhibir la proliferación y activación de fibroblastos en un modelo de ratón in vivo y la cicatriz hipertrófica humana in vitro (101). En la piel, se ha demostrado que la galangina se dirige selectivamente a la quinasa 5 similar al receptor de Activina (ALK5), un receptor I de TGF-β, lo que resulta en la regulación a la baja de la fosforilación de Smad2 y Smad3 de una manera dependiente de la dosis, sin alterar la expresión de Smad2, Smad3, Smad4, Smad6 y Smad7 totales (101). Esto conduce a la supresión de la translocación de Smad2/3 inducida por TGF-β1 en el núcleo, lo que resulta en la disminución de la expresión de los niveles de ARNm y proteínas de colágeno tipo I, colágeno tipo III y α-SMA (101). En resumen, la galangina se puede usar localmente en cicatrices cutáneas anormales (102).
Inhibidores de Enzima convertidora de angiotensina (IECA). La enzima convertidora de angiotensina (ECA) es un miembro esencial del sistema renina-angiotensina. La ECA media la conversión de angiotensina I en angiotensina II vasoconstrictora, cuya función es ayudar a mantener el volumen sanguíneo y la presión arterial (103). Estudios previos han indicado que la ECA es una enzima bioquímica crítica que está implicada en los procesos fibróticos de los órganos internos, incluida la fibrosis hepática (104, 105), la fibrosis renal y la fibrosis cardíaca (106). Curiosamente, también se ha demostrado que la ACE está involucrada en la formación de cicatrices. La fuente de ECA durante la formación de cicatrices se deriva tanto de la médula ósea como del tejido cutáneo (107).Los inhibidores de la enzima convertidora de angiotensina (IECA) se pueden usar para revertir el proceso fibrótico, ejemplificado por la atenuación de la remodelación ventricular con el tratamiento de ramipril (108), que es un tipo de IECA. Estudios recientes han sugerido que el IECA tiene un efecto inhibidor sobre la fibrosis de la piel (109, 110, 111). Tanto los estudios in vivo como in vitro han demostrado que el tratamiento con IECA da como resultado una disminución de la fosforilación de Smad2/3 y la quinasa1 activada por TGF-β(TAK1),una expresión educida de TGF-β1 y colágeno, y una proliferación suprimida de fibroblastos (110). En resumen, el IECA atenúa la cicatrización anormal en la piel al inhibir simultáneamente las vías Smad2/3 y TAK1. ACEI se usa comúnmente en la clínica en el sistema cardiovascular. Por lo tanto, el IECA puede ser un tratamiento seguro y eficaz para las cicatrices patológicas de la piel.
Posibles estrategias terapéuticas genéticas y celulares Ha habido una variedad de técnicas genéticas y celulares que se han investigado para prevenir la cicatrización. Se ha investigado la atenuación o el tratamiento de la cicatrización mediante ARN de interferencia (ARNi). ElARNi puede interferer con la transcripción de ciertos genes en fibroblastos y miofibroblastos, lo que podría eliminar la formación excesiva de colágeno, reduciendo potencialmente la cicatrización aberrante. Los ARN de interferencia pequeños (ARNip), ARN bicatenarios simétricos o asimétricos de aproximadamente 20 pares de bases, han recibido un interés notable debido a su capacidad para unirse al ARNmdiana complementario de una manera específica de secuencia a través del procesamiento de interferencia de ARN inherente (ARNi). ElARNip es responsible de la eliminación de genes específicos de la secuencia y la posterior suppression de la expression de proteínas, lo que resulta en un silenciamiento génico selective (112). Aunque el ARNip sintetizado químicamente se puede transfectar en células utilizando los reactivos de transfección, losARN de horquilla corta inductors de ARNi (ARNhc) se pueden administrar en las células utilizando vectores virales o no virales que codifican el ARNhc (113). Los micro ARN (miARN) son pequeños ARN no codificantes que se emparejan ampliamente con los ARNm de los genes codificantes de proteínas, lo que lleva a su repression postranscripcional (114). Y los miARN tienen un origen diferente y una conservación evolutiva distinta de los ARNip y el ARNhc (115). Los miARN que tienen menos de 200 nucleótidos (nt) de longitude son pequeños ARN no codificantes (ncRNA). Los ARN largos no codificantes (lncRNA) tienen una longitude superior a 200 nt y pueden ejercer efectos significativos sobre la transcripción a través de la interferencia, la remodelación de la cromatina y la regulación del empalme postranscripcional (116). A pesar de los resultados prometedores con las estrategias terapéuticas celulares y genéticas, la seguridad de estos métodos es cuestionable. Por ejemplo, se ha demostrado que inducen una respuesta inmune a través de la activación del receptor tipo toll (TLR) (112). Por lo tanto, se necesitan más estudios preclínicos y clínicos para evaluar más a fondo la eficacia y la seguridad de estos métodos.
- Interferencia de ARN por siRNA y shRNA
La subunidad A del factor de iniciación de la traducción eucariota 3 (eIF3a) es una de las subunidades centrales del complejo de iniciación de la traducción. Estudios anteriores han demostrado que la eliminación de eF3a puede inhibir la síntesis de colágeno en los fibroblastos renales a través de la inhibición de la vía de señalización TGF-β1/Smad (117). Sin embargo, el papel de eIF3a en la formación de queloides humanos sigue siendo desconocido. Cuando los fibroblastos queloides tratados con TGF-β1 recombinante humano se incuban con siRNA-eIF3a o siRNA-mock para inducer la eliminación de eIF3a, se inhibe la fosforilación de Smad2/3 y hay una disminución de la expression de α-SMA, colágenoI, TGF-βRI y TGF-βRII. Estos hallazgos sugieren que siRNA-eIF3a ejerce un efecto inhibidor sobre la vía de señalización TGF-β1/Smad en fibroblastos queloides (118).
La interferencia de ARN a través de siRNA también se puede utilizar para silenciar el factor regulador de interferón 3 (IRF3) y el dominio de union a nucleotides y la proteína de repetición C5 rica en leucina (NLRC5). IRF3 es un factor de transcripción que regula la inflamación, la proliferación y la respuesta inmune innata de las células (110,120) y está involucrado en la progression de la fibrosis cardiac (121). NLRC5 juega un papel crucial en la respuesta inmune innata (122,123) y regula la proliferación y activación inducida por TGF-β1 de las células estrelladas hepaticas durante la fibrosis hepatica (124). Curiosamente, sus efectos sobre los fibroblastos queloides siguen siendo desconocidos. Sin embargo, cuando los fibroblastos queloides tratados con TGF-β1recombinantehumanoseincubanconARNip-IRF3yARNip-NLRC5, exhiben una proliferación disminuida, una fosforilación suprimida de Smad2/3 y una expression reducida de α-SMA, colágeno I, TGF-βRI y TGF-βRII (125,126). Esto ocurre debido a un efecto inhibidor sobre la vía des eñalización TGF-β1/Smad a través del silenciamiento por ARNip (125,126). El ARNip también se puede usar para derribar el gen 1 temprano inducible por TGF-β(TIEG1), que puede promover la fosforilación de Smad2 al provocar un efecto inhibidor sobre el promoter Smad7 (127). La eliminación da como resultado una disminución de la expression y proliferación de colágeno, inhibe la migración e invasión de fibroblastos queloides y suprime la activación de la señalización de TGF-β1/Smad2 (127).
Otra aplicación novedosa de siRNA para atacar y silenciar el receptor TGF-β tipo I en fibroblastos cicatriciales hipertróficas humanos conduce a la inhibición de la fosforilación de las proteínas Smad y, por lo tanto, no se activan las vías de señalización TGF-β/Smad. Esto da como resultado una proliferación reducida de fibroblastos, una reducción de la deposición de ECM y una cicatrización atenuada de la herida (128).
La trombospondina-4 humana (TSP-4) media la angiogénesis inducida por TGF-β1, controla la remodelación tisular y puede eliminarse mediante ARNhc inductor de ARNi (129). La evidencial ha demostrado que existe una interacción mutual positive entre TSP-4 y TGF-β1, lo que sugiere que la expression de TSP-4 es promovida por TGF-β1 a través de Smad3 y p38 y, a su vez, TSP-4 regula positivamente la fosforilación de Smad3 y p38 (129). La eliminación de TSP-4 debería inhibir significativamente la formación de cicatrices hipertróficas a través del bloqueo de las vías TGF-β1/Smad3 y TGF-β1/p38.
- Interferencia de ARN por miRNA. La regulación postranscripcional de la expresión génica por miARN se puede utilizar para manipular la proliferación, la diferenciación y la apoptosis. Múltiples miARN participant en el proceso de formación anormal de cicatrices, cuyo resultado es la suppression de las bioactividades de fibroblastos o miofibroblastos. Esto se ejemplifica con miR-1224-5p, un supresor de tumores que promueve la apoptosis y suprime la proliferación e invasión de células de glioma maligno (130). Se ha demostrado que MiR-1224-5p ejerce un efecto inhibidor sobre los fibroblastos del queloide similar a un tumor a través del bloqueo de la vía de señalización TGF-β1/Smad3. Esto da como resultado una disminución de la proliferación e invasión de fibroblastos queloides y un aumento de la apoptosis de los fibroblastos queloides (131). Varios miARN específicos, incluidos miR-23a, miR-125b y miR-145, que se originan en exosomas derivados de células madre mesenquimales derivadas del cordon umbilical humano (uMSC-Exos) pueden bloquear la vía de señalización TGF-β/Smad2 y disminuir la expression de α-SMA. Esto conduce a la supresión de la diferenciación de miofibroblastos, la promoción del proceso de cicatrización de heridas y la formación de cicatrices aberrantes atenuadas (132). Algunos miARN pueden estimular la function fibroproliferativa de los fibroblastos.MiRNA-21, por ejemplo, promueve la expression de ECMyα-SMA inducida porTGF-β1 a través de la suppression de Smad7 en fibroblastos cicatriciales hipertróficos. Esto demuestra que miR-21 puede ser un objetivo crítico para el tratamiento de cicatrices hipertróficas (133). Se ha demostrado que el lncRNACOL1A2-AS1 actúa como un inhibidor competitivo que bloquea la unión de miR-21 a su gendiana, lo que resulta en la regulación positiva de Smad7 y una disminución de la proliferación de fibroblastos cicatriciales hipertróficos (134).
3) Transfección celular
La sobreexpresión de algunos genes también puede ejercer un efecto inhibidor sobre la vía de señalización TGF-β/Smad. Se ha informad oque la proteína relacionada con el factor de necrosis tumoral C1q6 (CTRP6), una nueva familia altamente conservada de parálogos de adiponectina, tiene una variedad de funciones ,incluidos efectos vasculoprotectores a través de la relajación vascular (135), funciones metabólicas a través de la regulación de hormonas (136), efectos inhibidores sobre la diferenciación de miofibroblastos y efectos inhibidores sobre la expresión de ECM después de un infarto de miocardio para mejorar la función cardíaca y atenuar la remodelación patológica (137).
Recientemente, un estudio ha demostrado que, cuando los fibroblastos dérmicos de la piel humana normal se transfectan con el vector pcDNA3.1 cargado con CTRP6, se inhibe la proliferación de fibroblastos dérmicos y se suprime la expresión de ECM, evitando el desarrollo de fibrosis cutánea (138). Se ha demostrado que CTRP6 inhibe la expresión de colágeno tipo I y α-SMA inducida por TGF-β1 y suprime la fosforilación de Smad3 (138). CTRP6 tiene baja expresión en tejidos cicatriciales y fibroblastos dérmicos tratados con TGF-β1, y no tiene ningún efecto sobre la fosforilación de Smad2 (138). CTRP6 puede ser un objetivo terapéutico potencial para la prevención de la fibrosis cutánea, pero se necesitan más estudios para dilucidar completamente su papel, ya que otros estudios han sugerido que CTRP6 está asociado con la obesidad y la inflamación del tejido adiposo y la resistencia a la insulina y está involucrado en el desarrollo de diabetes tipo II en la población china (139,140). Se ha demostrado que Dickkopf-3 (DKK3), un supresor de tumores que codifica proteínas secretadas, proporciona efectos beneficiosos significativos sobre las cicatrices queloides.
Cuando los fibroblastos queloides inducidos por TGF-β1 se transfectan con el vector pcDNA3.1-DKK3, se produce la apoptosis de los fibroblastos queloides, que se asocia con una mayor expresión de Bax y caspasa-3 y una disminución de lae xpresión de Bcl-2 (141). También se inhibe la expresión de los receptores TGF-βtipoI y tipo II y la fosforilación de Smad2 y Smad3 (141). Por lo tanto, DKK3 ejerce un efecto inhibidor sobre la vía de señalización TGF-β1/Smad en fibroblastos queloides, lo que sugiere que DKK3 puede ser un enfoque potencial en el tratamiento de la cicatrización queloide.
Estrategias terapéuticas combinadas prometedoras
La vía de señalización TGF-β/Smad es la vía canónica involucrada en la formación de colágeno en los fibroblastos y miofibroblastos, y se ha demostrado que múltiples estrategias terapéuticas, incluidas las terapias con medicamentos y los posibles métodos genéticos, inhiben esta vía. Sin embargo, la mayoría de estos estudios se han centrado en la monoterapia. La terapia génica combinada con nano partículas de oro conjugadas con polietilemina (PEI2-GNP) BMP-7 y factor de crecimiento de hepatocitos (HGF) tiene un efecto significativo en el tratamiento de la fibrosis corneal al inducir la apoptosis selectiva de miofibroblastos (142). Tanto BMP-7 como HGF pueden inducir una mayor expresión de caspasa-3 en el miofibroblasto (93,142). Estos resultados indican que un método de administración eficiente combinado con una terapia dual o incluso una terapia triple que se dirige a la vía de señalización TGF-β/Smad en los fibroblastos o miofibroblastos puede proporcionar un efecto inhibidor sinérgico significativo sobre laf ormación de cicatrices aberrantes en la piel. Por lo tanto, planteamos la hipótesis de que la administración de dos moléculas, como BMP-7 y DKK3, para tratar una cicatriz queloide puede inducir la apoptosis de los fibroblastos queloides, lo que resulta en la atenuación de cicatrices aberrantes.
Pirfenidona Tópica 8%
La pirfenidona es un fármaco antifibrótico aprobado por la FDA para usos sistémico en el tratamiento de la fibrosis pulmonar idiopática y otros trastornos fibróticos (137).
Por otro lado, la Pf tópica existe comercialmente en México en formato gel a una concentración del 8%.
Este gel de Pf se prescribe comúnmente para el tratamiento de heridas como las cirugías, quemaduras, dehiscencias y úlceras.
Se aplica de manera tópica sobre las zonas a tratar ya que los efectos adversos informados son menores que con la administración sistémica. La frecuencia de aplicación es dos veces por día en forma de capa delgada y la duración del tratamiento puede variar según la gravedad de las cicatrices y la respuesta individual al medicamento.
La utilización terapéutica de la Pf en forma tópica es promisoria dada los beneficios que ofrece la terapia, acortando tiempos de cicatrización o mejorando la cicatriz hipertrófica.
El mayor desafío será́ la realización de ensayos clínicos a mayor escala, para comprobar la calidad del epitelio obtenido con este tratamiento (138).
En resumen:
- Fácil aplicación tópica.
- Minimización de la apariencia de cicatrices: puede ser reduciendo la pigmentación, suavizando la textura o promoviendo la regeneración celular, muy importante en cicatrices visibles, como las causadas por cirugías o quemaduras.
- Menor probabilidad de efectos secundarios sistémicos, por su aplicación tópica, limitando su acción en la zona de aplicación.
- Resultados Variables: La eficacia de los geles tópicos para cicatrices puede variar según la persona y el tipo de herida. Algunas personas pueden experimentar una mejora significativa, mientras que en otros casos los resultados pueden ser más modestos.
- Tiempo y Paciencia: Los resultados no son inmediatos y pueden requerir semanas o incluso meses de uso constante. La paciencia y la adherencia al tratamiento son clave para obtener beneficios significativos.
- Costo y Accesibilidad:este tipo de geles pueden ser costosos y su disponibilidad puede variar según la ubicación geográfica, pudiendo afectar la accesibilidad para ciertos individuos.
CONCLUSIONES
Aunque la formación de cicatrices es un sistema de reparación eficaz para la evolución genética humana, una vez que se produce una cicatriz, no se puede eliminar por completo. Los tejidos cicatriciales ocupan las estructuras de los tejidos normales originales, afectando gravemente sus funciones normales y estética. Las terapias que pueden restaurar la función y la apariencia del tejido dañado son de gran interés. En la actualidad, la profilaxis y el tratamiento de las cicatrices son una preocupación mundial. En general, las opciones de tratamiento para las cicatrices se pueden dividir en tres clases, ya sea tratamiento quirúrgico, tratamiento no quirúrgico o la combinación de ambos (9).
La formación de cicatrices fibróticas aberrantes es el resultado final del proceso de cicatrización de heridas patológicamente desorganizado. Por lo tanto, interferir con este proceso puede reducir la fibrosis hasta cierto punto, evitando así la fibrosis excesiva. Se ha logrado un gran progreso en la comprensión de los mecanismos básicos de cómo los fibroblastos y los miofibroblastos leen la señalización de TGF-β/Smad en condiciones patológicas y producen una gran cantidad de colágenos desordenados que conducen a la formación de cicatrices aberrantes. Sin embargo, actualmente no hay fármacos disponibles para la inhibición completa de la fibrosis, y todavía existen limitaciones para el tratamiento estándar de las cicatrices aberrantes. Dado que se ha demostrado que las monoterapias múltiples ejercen efectos inhibidores sobre la vía de señalización canónica TGF-β/Smad y se ha informado que las terapias combinadas prometedoras inhiben significativamente la fibrosis en otros órganos, planteamos la hipótesis de que la combinación de algunos de los tratamientos de monoterapia mencionados anteriormente puede tener un efecto inhibidor sinérgico notable sobre la formación anormal de cicatrices en la piel. Sin embargo, no importa cuán artificialmente intervenga en el proceso de formación de cicatrices, el grado de fibrosis solo se puede atenuar posiblemente en la mayor medida posible, y actualmente no se puede lograr la curación sin cicatrices.
El gel de Pirfenidona ofrece una opción tópica efectiva para mejorar la apariencia de cicatrices. Sin embargo, su uso debe ser supervisado por un profesional de la salud, y es esencial seguir las indicaciones para maximizar los beneficios y minimizar los posibles efectos secundarios. Este trabajo proporciona una visión general del uso de la Pf en gel para cicatrices. El objetivo será que en nuestro país se logren realizar ensayos clínicos para poder comercializarlo, por todas las ventajas descriptas, pudiendo ser una opción alternativa incruenta, evitando tratamiento quirúrgico.
Los estudios han demostrado que la piel fetal lesionada se puede regenerar al principio del embarazo sin la formación de tejido cicatricial (145), lo que sugiere que es posible una curación sin cicatrices. La curación sin cicatrices implicala regeneración celular, en lugar de la reparación. Los numerosos factores de crecimiento y células madre presentes en el líquido amniótico humano proporcionan una condición indispensable para la regulación precisa de la regeneración celular (146, 147, 148). Aunque se requieren más estudios para comprender mejor este proceso, se puede lograr una curación sin cicatrices proporcionando un microambiente similar al del líquido amniótico.
ANEXOS

Fig. 1. Remodelación de la herida y los dos tipos de resultados. Laformación de cicatrices es un resultado inevitable después de la lesión en la piel. Se espera que las posibles estrategias terapéuticas, incluidas las estrategias terapéuticas con medicamentos y las estrategias terapéuticas genéticas, atenúen la formación de cicatrices aberrantes.

Fig. 2. Vía dependiente de Smad inducida por TGF-β. El resultado de la unión del ligando y el receptor es la activación del receptor tipo II, que luego se permite fosforilar el receptor tipo I. Después de la fosforilación del receptor tipo I, reconoce y fosforila específicamente los R-Smads, incluidos Smad2 y Smad3, y la fosforilación de ambos desestabiliza la interacción con el anclaje Smad para la activación del receptor (SARA) y aumenta la afinidad por Smad4 (el llamado Co-Smad). Cuando Smad2 y Smad3 se liberan de SARA, pueden interactuar con Smad4 para formar un complejo transcripcional, que es libre de translocarse al núcleo, unirse a coactivadores o correpresores transcripcionales y regular la actividad transcripcional de varios genes. Además, Smad2 y Smad3 también son reconocidos por los receptores de activina y ganglionar estrechamente relacionados. Otros R-Smads como Smad1, Smad5 y Smad8 son reconocidos principalmente por BMP. Las ubiquitinaligasas E3 Smad factor relacionado con la ubiquitina-1 (Smurf1) y Smurf2 son reclutadas por Smad7, Smurf1 interactúa selectivamente con Smads 1, 5 y 8 en el citoplasma para desencadenar su ubiquitinación, Smurf2 se dirige selectivamente al Smad2 activado en el núcleo para su degradación, mientras que la ubiquitinación de Smad3 en el núcleo está mediada por un complejo diferente de ubiquitinaligasa E3 ROC1-SCFFbw1a. El complejo Pitufo y Smad7 media la ubiquitinación y la degradación mediada por proteasoma del receptor TGF-β tipo I. Los resultados anteriores conducen a la terminación de la señalización TGF-β/Smad.

Fig. 3. Los efectos de todas las estrategias terapéuticas dirigidas a la vía de señalización TGF-β/Smad, incluida la proliferación inhibida de fibroblastos, promovieron la apoptosis de fibroblastos, bloquearon la diferenciación de fibroblastos en miofibroblastos y suprimieron la síntesis de colágeno.
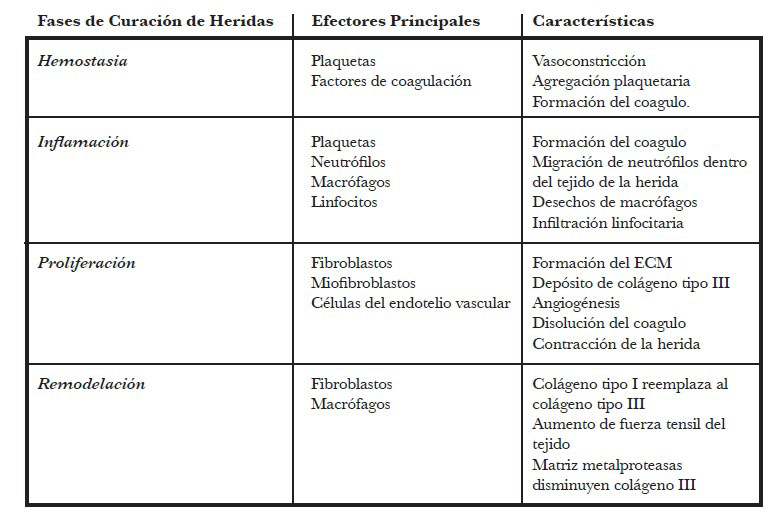 Tab. 1
Tab. 1
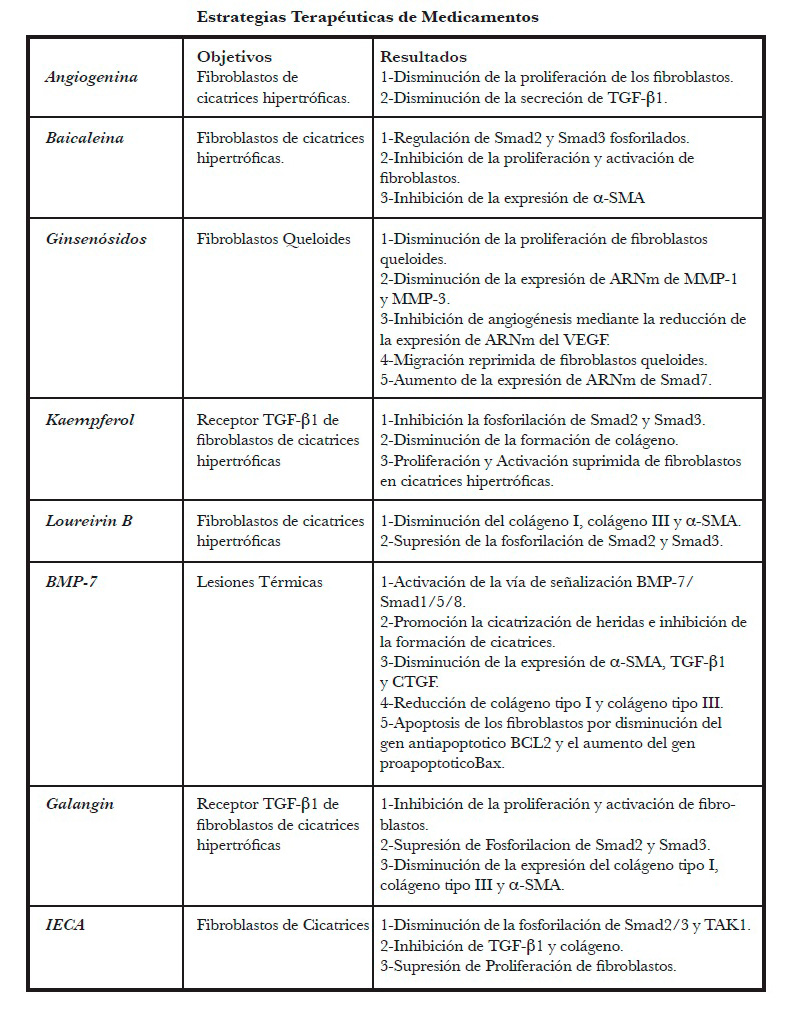
Tab. 2

Tab. 3
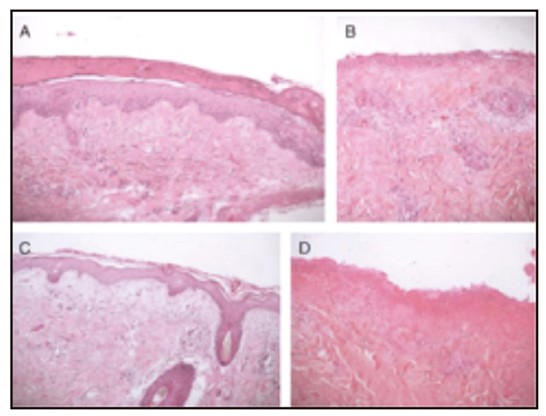
Biopsia de piel, día 7.
A y C grupo tratado con pirfenidona.
B y D grupo tratado con tratamiento convencional.

Evaluación clínica, día 7.
A y C grupo tratado con pirfenidona.
B y D grupo tratado con tratamiento convencional.


Bibliografía
-
Balaraman, E.R. Geddes, P.M. Friedman. Best reconstructive techniques Dermatol. Surg., 41 (2015), pp. S265-S275.
-
Martin, R. Nunan. Cellular and molecular mechanisms of repair in acute and chronic wound healingBr. J. Dermatol., 173 (2) (2015), pp. 370-378,
-
Satish L, Evdokiou A, Geletu E, Hahn JM, and Supp D. Pirfenidone inhibits epithelial–mesenchymal transition in keloid keratinocytes. Burns & Trauma, 2020, 8, tkz007 doi: 10.1093/burnst/tkz007 Research Article.
-
Chung EP, Nguyen J, Tellkamp-Schehr T, Goebel K, Ollek A, Krein C, et al. A Soft Skin Adhesive (SSA) Patch for Extended Release of Pirfenidone in Burn Wounds. 2023, 15, 1842. https://doi.org/10.3390/ pharmaceutics15071842.
-
Wells AR, Leung K. Pirfenidone attenuates the profibrotic contractile phenotype of differentiated human dermal myofibroblasts. © 2019 The Authors. Published by Elsevier Inc. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
-
Medina J, Sebastian EA, Fourcaudot A, Dorati R, and Leung K. Pirfenidone Ointment Modulates the Burn Wound Bed in C57BL/6 Mice by Suppressing Inflammatory Responses. 0360-3997/18/0000-0001/0 # 2018 Springer Science+Business Media, LLC, part of Springer Nature.DOI: 10.1007/s10753-018-0871-y.
-
Mecott-Rivera GA, Aguilar-Baqueiro JA, Bracho S, Miranda-Maldonado I, Franco-Márquez R, Castro-Govea Y, et al. Pirfenidone increases the epithelialization rate of skin graft donor sites. JBUR 5597 No. of Pages 8.
-
Mecott GA, MMS; González-Cantú I, MD; Dorsey-Treviño EG, MD; Matta-Yee-Chig D, MSC; Saucedo-Cárdenas O, PhD; Montes de Oca-Luna R, PhD, et al. Efficacy and Safety of Pirfenidone in Patients with Second-Degree Burns: A Proof-of-Concept Randomized .Controlled Trial Adv Skin Wound Care 2020;33:1–7. DOI: 10.1097/01.ASW.0000655484.95155.f7.
-
Yagmur, et al. Mechanical receptor-related mechanisms in scar management: a review and hypothesis Plast. Reconstr. Surg., 126 (2) (2010), pp. 426-434.
-
T. Robles, D. Berg. Abnormal wound healing: keloidsClin. Dermatol., 25 (1) (2007), pp. 26-32.
-
G. Gauglitz, et al. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategiesMol Med, 17 (1-2) (2011), pp. 113-125.
-
Bataller, D.A. Brenner. Liver fibrosis J. Clin. Invest., 115 (2) (2005), pp. 209-218.
-
A. Wynn, T.R. Ramalingam. Mechanisms of fibrosis: therapeutic translation for fibrotic disease Nat. Med., 18 (7) (2012), pp. 1028-1040.
-
K. Lichtman, M. Otero-Vinas, V. Falanga. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosisWound Repair Regen., 24 (2) (2016), pp. 215-222,
-
Pohlers, et al. TGF-β and fibrosis in different organs — molecular pathway imprintsBBA-Mol Basis Dis., 1792 (8) (2009), pp. 746-756.
-
M.-K. Tang, et al. Transforming growth factor-βsignalling in renal fibrosis: from Smads to non-coding RNAsJ. Physiol. (Lond.), 596 (16) (2018), pp. 3493-3503.
-
M. Carthy, TGFbeta. Signaling and the control of myofibroblast differentiation: implications for chronic inflammatory disorders J. Cell. Physiol., 233 (1) (2018), pp. 98-106.
-
L. Moses, A.B. Roberts, R. Derynck. The discovery and early days of TGF-β: a historical perspectiveCold Spring Harb. Perspect.Biol., 8 (7) (2016).
-
G. Gauglitz. Management of keloids and hypertrophic scars: current and emerging options Clin. Cosmet. Investig. Dermatol., 6 (2013), pp. 103-114.
-
S, H.G.B.M.S.H.M.M.F.R.WDifferential regulation of pro-inflammatory cytokines during wound healing in normal and glucocorticoid-treated miceCytokine, 8 (7) (1996), pp. 548-556.
-
Guo, L.A. Dipietro. Factors affecting wound healing J. Dent. Res., 89 (3) (2010), pp. 219-229.
-
W. Segal. How neutrophils kill microbesAnnu. Rev. Immunol., 23 (2005), pp. 197-223.
-
Mahdavian Delavary, et al. Macrophages in skin injury and repair Immunobiology, 216 (7) (2011), pp. 753-762.
-
O. Martinez, S. Gordon. The M1 and M2 paradigm of macrophage activation: time for reassessment F1000Prime Rep., 6 (2014).
-
Sica, A. Mantovani. Macrophage plasticity and polarization: in vivo veritas J. Clin. Invest., 122 (3) (2012), pp. 787-795.
-
Wild, et al. Basics in nutrition and wound healingNutrition, 26 (9) (2010), pp. 862-866.
-
H. Sun, et al. Alpha-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFbeta activation or collagen production across multiple models of organ fibrosisAm. J. Physiol. Lung Cell Mol. Physiol., 310 (9) (2016), pp. L824-836.
-
Huet, et al. Extracellular matrix metalloproteinase inducer/CD147 promotes myofibroblast differentiation by inducing alpha-smooth muscle actin expression and collagen gel contraction: implications in tissue remodeling FASEB J., 22 (4) (2008), pp. 1144-1154.
-
Hinz, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling Am. J. Pathol., 180 (4) (2012), pp. 1340-1355.
-
C. Gurtner, et al. Wound repair and regeneration Nature, 453 (7193) (2008), pp. 314-321.
-
A. O’Toole. Extracellular matrix and keratinocyte migration Clin. Exp. Dermatol., 26 (6) (2001), pp. 525-530.
-
J. Baryza, G.A. Baryza. The Vancouver Scar Scale: an administration tool and its interrater reliability J. Burn Care Rehabil., 16 (5) (1995), pp. 535-538.
-
Neri, et al. Ischemia/Reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists Mediators Inflamm., 2017 (2017), p. 7018393.
-
Gabbiani. The myofibroblast in wound healing and fibrocontractive diseases J. Pathol., 200 (4) (2003), pp. 500-503.
-
Kalluri, M. Zeisberg Fibroblasts in cancer Nat. Rev. Cancer, 6 (5) (2006), pp. 392-401.
-
Desmouliere, et al. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts Cell Biol., 122 (1) (1993), pp. 103-111.
-
H. Heldin, K. Miyazono, P. ten Dijke. TGF-beta signalling from cell membrane to nucleus through SMAD proteins Nature, 390 (6659) (1997), pp. 465-471.
-
Miyazono, S. Maeda, T. ImamuraBMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talkCytokine Growth Factor Rev., 16 (3) (2005), pp. 251-263,
-
Ikushima, K. Miyazono. TGF-beta signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-betaCell Tissue Res., 347 (1) (2012), pp. 37-49,
-
Shi, J. Massagué. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus Cell, 113 (6) (2003), pp. 685-700.
-
J. You, et al. The type III TGF-beta receptor signals through both Smad3 and the p38 MAP kinase pathways to contribute to inhibition of cell proliferation Carcinogenesis, 28 (12) (2007), pp. 2491-2500.
-
Chen, et al. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signalingScience, 301 (5638) (2003), pp. 1394-1397.
-
C. Kirkbride, et al. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptorJ. Biol. Chem., 283 (12) (2008), pp. 7628-7637.
-
A. Lewis, et al. Beta glycan binds inhibin and can mediate functional antagonism of activin signallingNature, 404 (6776) (2000), pp. 411-414.
-
Wiater, et al. Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actionsJ. Biol. Chem., 281 (25) (2006), pp. 17011-17022.
-
Massague. How cells read TGF-beta signalsNat. Rev. Mol. Cell Biol., 1 (3) (2000), pp. 169-178.
-
Hu, Z. Wu, S.H. Phan. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression Am. J. Respir. Cell Mol. Biol., 29 (3 Pt 1) (2003), pp. 397-404.
-
-J. Chen, et al. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-β: involvement of smad 3J. Invest. Dermatol., 112 (1) (1999), pp. 49-57.
-
Verrecchia, et al. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific mannerOncogene, 20 (26) (2001), pp. 3332-3340.
-
K. Ghosh, et al. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators Oncogene, 19 (31) (2000), pp. 3546-3555.
-
Hata, Y.G. Chen. TGF-beta signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol., 8 (9) (2016).
-
Hayashi, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling Cell, 89 (7) (1997), pp. 1165-1173.
-
Imamura, et al. Smad6 inhibits signalling by the TGF-beta superfamily Nature, 389 (6651) (1997), pp. 622-626.
-
S. Lo, J. Massague. Ubiquitin-dependent degradation of TGF-beta-activated smad2 Nat. Cell Biol., 1 (1999), pp. 472-478.
-
Zhu, et al.. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation Nature, 400 (6745) (1999), pp. 687-693.
-
Lin, M. Liang, X.-H. Feng. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth Factor-β signaling J. Biol. Chem., 275 (47) (2000), pp. 36818-36822.
-
Fukuchi, et al. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins Mol. Biol. Cell, 12 (2001), pp. 1431-1443.
-
Kavsak, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation Mol. Cell, 6 (6) (2000), pp. 1365-1375.
-
Miyazawa, K. Miyazono. Regulation of TGF-β family signaling by inhibitory smads Cold Spring Harb. Perspect.Biol., 9 (3) (2017).
-
Suzuki, et al. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane J. Biol. Chem., 277 (42) (2002), pp. 39919-39925.
-
Yan, X. Xiong, Y.G. Chen. Feedback regulation of TGF-beta signaling Acta BiochimBiophys Sin (Shanghai), 50 (1) (2018), pp. 37-50.
-
Gao, Z. Xu. Mechanisms of action of angiogeninActa. Biochim. Biophys. Sin (Shanghai), 40 (7) (2008), pp. 619-624.
-
C. Pan, et al. Angiogenin expression in burn blister fluid: implications for its role in burn wound neovascularization Wound Repair Regen., 20 (5) (2012), pp. 731-739.
-
Skorupa, et al. Angiogenin induces modifications in the astrocyte secretome: relevance to amyotrophic lateral sclerosis J. Proteomics, 91 (2013), pp. 274-285.
-
C. Pan, et al. Angiogenin attenuates scar formation in burn patients by reducing fibroblast proliferation and transforming growth factor beta1 secretion Ann. Plast. Surg., 80 (2S Suppl 1) (2018), pp. S79-S83.
-
Bie, et al. Baicalein: a review of its anti-cancer effects and mechanisms in Hepatocellular Carcinoma Biomed. Pharmacother., 93 (2017), pp. 1285-1291.
-
Li, J. Zhao, C. Holscher. Therapeutic potential of Baicalein in alzheimer’s disease and parkinson’s disease CNS Drugs, 31 (8) (2017), pp. 639-652.
-
Sowndhararajan, et al. Baicalein as a potent neuroprotective agent: a review Biomed. Pharmacother., 95 (2017), pp. 1021-1032.
-
-G. Park, et al. Baicalein inhibits tumor progression by inhibiting tumor cell growth and tumor angiogenesis Oncol. Rep., 38 (5) (2017), pp. 3011-3018.
-
J. Chen, et al. Scutellariae radix suppresses LPS-induced liver endothelial cell activation and inhibits hepatic stellate cell migration J. Ethnopharmacol., 150 (3) (2013), pp. 835-842.
-
K. Kong, et al. Baicalein and Wogonin inhibit collagen deposition in SHR and WKY cardiac fibroblast cultures BMB Rep., 43 (4) (2010), pp. 297-303.
-
S. Patwardhan, et al. Baicalein exhibits anti-inflammatory effects via inhibition of NF-kappaB transactivation Biochem. Pharmacol., 108 (2016), pp. 75-89.
-
Miyazono, Y. Kamiya, M. Morikawa. Bone morphogenetic protein receptors and signal transduction J. Biochem., 147 (1) (2010), pp. 35-51
-
F. Zhang, et al. Baicalein attenuates hypertrophic scar formation via inhibition of the transforming growth factor-β/Smad2/3 signalling pathway Br. J. Dermatol., 174 (1) (2016), pp. 120-130.
-
C. He, et al. Ginsenoside Rg3 inhibits colorectal tumor growth through the down-regulation of Wnt/ss-catenin signalingInt. J. Oncol., 38 (2) (2011), pp. 437-445.
-
Yang, et al. Ginsenoside Rg3 inhibits colorectal tumor growth via down-regulation of C/EBPbeta/NF-kappaB signaling Biomed. Pharmacother., 96 (2017), pp. 1240-1245.
-
H. Wang, et al. 20(s)-ginsenoside Rg3 promotes apoptosis in human ovarian cancer HO-8910 cells through PI3K/Akt and XIAP pathways Tumour Biol., 35 (12) (2014), pp. 11985-11994.
-
Tang, et al. Ginsenoside Rg3 inhibits keloid fibroblast proliferation, angiogenesis and collagen synthesis in vitro via the TGFbeta/Smad and ERK signaling pathways Int. J. Mol. Med., 41 (3) (2018), pp. 1487-1499.
-
Fujiwara, Y. Muragaki, A. Ooshima. Keloid-derived fibroblasts show increased secretion of factors involved in collagen turnover and depend on matrix metalloproteinase for migration Br. J. Dermatol., 153 (2) (2005), pp. 295-300.
-
Uchida, et al. Tretinoin reverses upregulation of matrix metalloproteinase-13 in human keloid-derived fibroblasts Exp. Dermatol., 12 (Suppl 2) (2003), pp. 35-42.
-
P. Devi, et al. Kaempferol and inflammation: from chemistry to medicine Pharmacol. Res., 99 (2015), pp. 1-10.
-
F. Lee, et al. Kaempferol induces ATM/p53-mediated death receptor and mitochondrial apoptosis in human umbilical vein endothelial cellsInt. J. Oncol., 48 (5) (2016), pp. 2007-2014.
-
Maini, B.M. Fahlman, E.S. Krol. Flavonols protect against UV radiation-induced thymine dimer formation in an artificial skin mimic J. Pharm. Pharm. Sci., 18 (4) (2015), pp. 600-615.
-
Li, et al. Kaempferol inhibits fibroblast collagen synthesis, proliferation and activation in hypertrophic scar via targeting TGF-β receptor type I Biomed. Pharmacother., 83 (2016), pp. 967-974.
-
Liu, et al. Evaluation of the wound healing potential of Resina draconis (Dracaena cochinchinensis) in animal models Evid. Based Complement. Altern. Med., 2013 (2013), pp. 1-10.
-
Bai, et al. Loureirin B inhibits fibroblast proliferation and extracellular matrix deposition in hypertrophic scar via TGF-beta/Smad pathway Exp. Dermatol., 24 (5) (2015), pp. 355-360.
-
R. Bi, et al. The ratio of transforming growth factor-β1/bone morphogenetic protein-7 in the progression of the epithelial-mesenchymal transition contributes to rat liver fibrosisGenet. Med. Res., 13 (1) (2014), pp. 1005-1014.
-
Merino, et al. BMP-7 attenuates left ventricular remodelling under pressure overload and facilitates reverse remodelling and functional recovery Cardiovasc. Res., 110 (3) (2016), pp. 331-345.
-
Urbina, D.K. Singla. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy Am. J. Physiol. Heart Circ. Physiol., 307 (5) (2014), pp. H762-772.
-
F. Higgins, et al. BMP7-induced-Pten inhibits Akt and prevents renal fibrosisBBA-Mol Basis Dis., 1863 (12) (2017), pp. 3095-3104.
-
P. Wang, et al. BMP-7 attenuates liver fibrosis via regulation of epidermal growth factor receptor Int. J. Clin. Exp. Pathol., 7 (7) (2014), pp. 3537-3547.
-
Liang, et al. BMP-7 attenuated silica-induced pulmonary fibrosis through modulation of the balance between TGF-beta/Smad and BMP-7/Smad signaling pathwayChem. Biol. Interact., 243 (2016), pp. 72-81.
-
Guo, et al. BMP-7 suppresses excessive scar formation by activating the BMP-7/Smad1/5/8 signaling pathway Mol. Med. Rep., 16 (2) (2017), pp. 1957-1963.
-
Wang, et al. Galangin suppresses hepatocellular carcinoma cell proliferation by reversing the Warburg effect Biomed. Pharmacother., 95 (2017), pp. 1295-1300.
-
W. Zou, S.P. Xu. Galangin inhibits the cell progression and induces cell apoptosis through activating PTEN and Caspase-3 pathways in retinoblastoma Biomed. Pharmacother., 97 (2018), pp. 851-863.
-
X. Wang, C. Tang. Galangin suppresses human laryngeal carcinoma via modulation of caspase-3 and AKT signaling pathways Oncol. Rep., 38 (2) (2017), pp. 703-714.
-
Yang, et al. Galangin suppresses human osteosarcoma cells: an exploration of its underlying mechanism Oncol. Rep., 37 (1) (2017), pp. 435-441.
-
C. Huang, et al. Galangin ameliorates cisplatin-induced nephrotoxicity by attenuating oxidative stress, inflammation and cell death in mice through inhibition of ERK and NF-kappaB signaling Toxicol. Appl. Pharmacol., 329 (2017), pp. 128-139.
-
Y. Wen, et al. Galangin suppresses H2 O2 -induced aging in human dermal fibroblasts Environ. Toxicol., 32 (12) (2017), pp. 2419-2427.
-
J. Choi, et al. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: critical role of PPAR-gamma signaling pathway Biochem. Pharmacol., 144 (2017), pp. 120-131.
-
Zhang, et al. Galangin inhibits hypertrophic scar formation via ALK5/Smad2/3 signaling pathway Mol. Cell. Biochem., 413 (1-2) (2016), pp. 109-118.
-
K. Kwak, et al. Galangin enhances TGF-beta1-mediated growth inhibition by suppressing phosphorylation of threonine 179 residue in Smad3 linker region Biochem. Biophys. Res. Commun., 494 (3-4) (2017), pp. 706-713.
-
E. Bernstein, et al. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme Pharmacol. Rev., 65 (1) (2013), pp. 1-46.
-
Noguchi, et al. Serum angiotensin-converting enzyme level for evaluating significant fibrosis in chronic hepatitis B World J. Gastroenterol., 23 (36) (2017), pp. 6705-6714.
-
S.A.C. Simoes, M.M. Teixeira. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis Pharmacol. Res., 107 (2016), pp. 154-162.
-
S. Miranda, E.S.A.C. Simoes. Serum levels of angiotensin converting enzyme as a biomarker of liver fibrosis World J. Gastroenterol., 23 (48) (2017), pp. 8439-8442.
-
Q. Fang, et al. The source of ACE during scar formation is from both bone marrow and skin tissue FASEB J., 32 (9) (2018), pp. 51995208.
-
Wei, et al. Ramipril attenuates left ventricular remodeling by regulating the expression of activin A-follistatin in a rat model of heart failure Sci. Rep., 6 (2016), p. 33677.
-
Q. Fang, et al. Angiotensin-converting enzyme inhibitor reduces scar formation by inhibiting both canonical and noncanonical TGF-beta1 pathways Sci. Rep., 8 (1) (2018), p. 3332.
-
Q. Tan, et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-beta-activated kinase 1 (TAK1) pathways in mice Br. J. Pharmacol., 175 (22) (2018), pp. 4239-4252.
-
Zheng, et al. The effect of topical ramipril and losartan cream in inhibiting scar formation Biomed. Pharmacother., 118 (2019), p. 109394.
-
H. Lee, et al. Current preclinical small interfering RNA (siRNA)-based conjugate systems for RNA therapeutics Adv. Drug Deliv. Rev., 104 (2016), pp. 78-92.
-
Machitani, et al. Type I interferons impede short hairpin RNA-Mediated RNAi via inhibition of dicer-mediated processing to small interfering RNA Mol. Ther. €” Nucleic Acids, 6 (2017), pp. 173-182.
-
P. Bartel. Metazoan MicroRNAs Cell, 173 (1) (2018), pp. 20-51.
-
P. Bartel. MicroRNAs: genomics, biogenesis, mechanism, and function Cell, 116 (2) (2004), pp. 281-297.
-
Ma, V.B. Bajic, Z. Zhang- On the classification of long non-coding RNAs RNA Biol., 10 (6) (2014), pp. 924-933.
-
F. Zhang, et al. Knockdown of elF3a inhibits collagen synthesis in renal fibroblasts via Inhibition of transforming growth factor-beta1/Smad signaling pathway Int. J. Clin. Exp. Pathol., 8 (2015), pp. 8983-8989.
-
Li, J. Zhao. Knockdown of elF3a inhibits TGFbeta1induced extracellular matrix protein expression in keloid fibroblasts Mol. Med. Rep., 17 (3) (2018), pp. 4057-4061.
-
Kumari, et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning J. Clin. Invest., 126 (8) (2016), pp. 2839-2854.
-
L. Tian, et al. IRF3 is involved in human acute myeloid leukemia through regulating the expression of miR-155 Biochem. Biophys. Res. Commun., 478 (3) (2016), pp. 1130-1135.
-
Tsushima, et al. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension FASEB J., 25 (5) (2011), pp. 1531-1543.
-
Luan, et al. NLRC5 inhibits neointima formation following vascular injury and directly interacts with PPARgamma Nat. Commun., 10 (1) (2019), p. 2882.
-
Wang, et al. NLRC5 negatively regulates LTA-induced inflammation via TLR2/NF-kappaB and participates in TLR2-mediated allergic airway inflammationJ. Cell. Physiol., 234 (11) (2019), pp. 19990-20001.
-
Xu, et al. NLRC5 regulates TGF-beta1-induced proliferation and activation of hepatic stellate cells during hepatic fibrosisInt. J. Biochem. Cell Biol., 70 (2016), pp. 92-104.
-
L. Ma, et al. Silencing NLRC5 inhibits extracellular matrix expression in keloid fibroblasts via inhibition of transforming growth factor-beta1/Smad signaling pathway Biomed. Pharmacother., 83 (2016), pp. 1016-831021.
-
Zhang, et al. Knockdown of IRF3 inhibits extracellular matrix expression in keloid fibroblasts. Biomed. Pharmacother., 88 (2017), pp. 1064-1068.
-
C. Hu, et al. TIEG1 represses Smad7-Mediated activation of TGF-beta1/Smad signaling in Keloid Pathogenesis J. Invest. Dermatol., 137 (5) (2017), pp. 1051-1059.
-
W. Wang, et al. siRNA-targeting transforming growth factor-beta type I receptor reduces wound scarring and extracellular matrix deposition of scar tissue J. Invest. Dermatol., 134 (7) (2014), pp. 2016-2025.
-
Qian, et al. Thrombospondin-4 critically controls transforming growth factor beta1 induced hypertrophic scar formationJ. Cell. Physiol., 234 (1) (2018), pp. 731-739.
-
Qian, et al. MiR-1224-5p acts as a tumor suppressor by targeting CREB1 in malignant gliomas Mol. Cell. Biochem., 403 (1-2) (2015), pp. 33-41.
-
Yao, et al. Tumor suppressive role of miR-1224-5p in keloid proliferation, apoptosis and invasion via the TGF-beta1/Smad3 signaling pathwayBiochem. Biophys. Res. Commun., 495 (1) (2018), pp. 713-720.
-
Fang, et al. Umbilical cord-derived mesenchymal stem cell-derived exosomal MicroRNAs suppress myofibroblast differentiation by inhibiting the transforming growth Factor-β/SMAD2 pathway during wound healing Stem Cells Transl. Med., 5 (10) (2016), pp. 1425-1439.
-
Li, et al. Fibroproliferative effect of microRNA-21 in hypertrophic scar derived fibroblasts Exp. Cell Res., 345 (1) (2016), pp. 93-99.
-
Nong, et al. LncRNA COL1A2-AS1 inhibits the scar fibroblasts proliferation via regulating miR-21/Smad7 pathway Biochem. Biophys. Res. Commun., 495 (1) (2018), pp. 319-324.
-
Zheng, et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathwayArterioscler. Thromb. Vasc. Biol., 31 (11) (2011), pp. 2616-2623.
-
M. Seldin, S.Y. Tan, G.W. Wong. Metabolic function of the CTRP family of hormones Rev. Endocr. Metab. Disord., 15 (2) (2013), pp. 111-123.
-
Shaojun Shi, MD, Jianhong Wu, MD, Huating Chen, BS, Hui Chen, MS, Jun Wu, MD, and Fandian Zeng, MD. Single- and Multiple-Dose Pharmacokinetics of Pirfenidone, an Antifibrotic Agent, in Healthy Chinese Volunteers. Journal of Clinical Pharmacology, 2007;47:1268-1276 © 2007 the American College of Clinical Pharmacology.
-
Bombaro K, Engrav L, Carrougher G, Wiechman S, Faucher L, Costa B, et al. What is the prevalence of hypertrophic scarring following burns? 0305-4179/03/$30.00 © 2003 Elsevier Science Ltd and ISBI. doi:10.1016/S0305-4179(03)00067-6.
-
Lei, et al. C1q/tumor necrosis factor-related protein-6 attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF-A pathway and inhibiting myofibroblast differentiation Basic Res. Cardiol., 110 (4) (2015), p. 35.
-
H. Fan, et al. CTRP6 inhibits fibrogenesis in TGF-beta1-stimulated human dermal fibroblastsBiochem. Biophys. Res. Commun., 475 (4) (2016), pp. 356-360.
-
Lei, et al. C1q/TNF-related protein 6 (CTRP6) links obesity to adipose tissue inflammation and insulin resistance J. Biol. Chem., 292 (36) (2017), pp. 14836-14850.
-
Wang, et al. C1q/TNF-related protein-6 is associated with insulin resistance and the development of diabetes in Chinese population Acta Diabetol., 55 (12) (2018), pp. 1221-1229.
-
Li, et al. DKK3 regulates cell proliferation, apoptosis and collagen synthesis in keloid fibroblasts via TGF-β1/Smad signaling pathway Biomed. Pharmacother., 91 (2017), pp. 174-180.
-
Gupta, et al. Novel combination BMP7 and HGF gene therapy instigates selective myofibroblast apoptosis and reduces corneal haze in vivoInvestig. Opthalmology Vis. Sci., 59 (2) (2018).
-
Rowlatt. Intrauterine wound healing in a 20 week human fetusVirchows. Arch. A Pathol. Anat. Histol., 381 (3) (1979), pp. 353-361.
-
Bajek, et al. Human amniotic-fluid-derived stem cells: a unique source for regenerative medicine. Expert Opin. Biol. Ther., 14 (6) (2014), pp. 831-839.
-
Hirai, et al. Trophic effect of multiple growth factors in amniotic fluid or human milk on cultured human fetal small intestinal cells. J. Pediatr. Gastroenterol. Nutr., 34 (5) (2002), pp. 524-528.
-
Joo, et al. Amniotic fluid-derived stem cells in regenerative medicine research. Arch. Pharm. Res., 35 (2) (2012), pp. 271-280.